Dioxygen activation
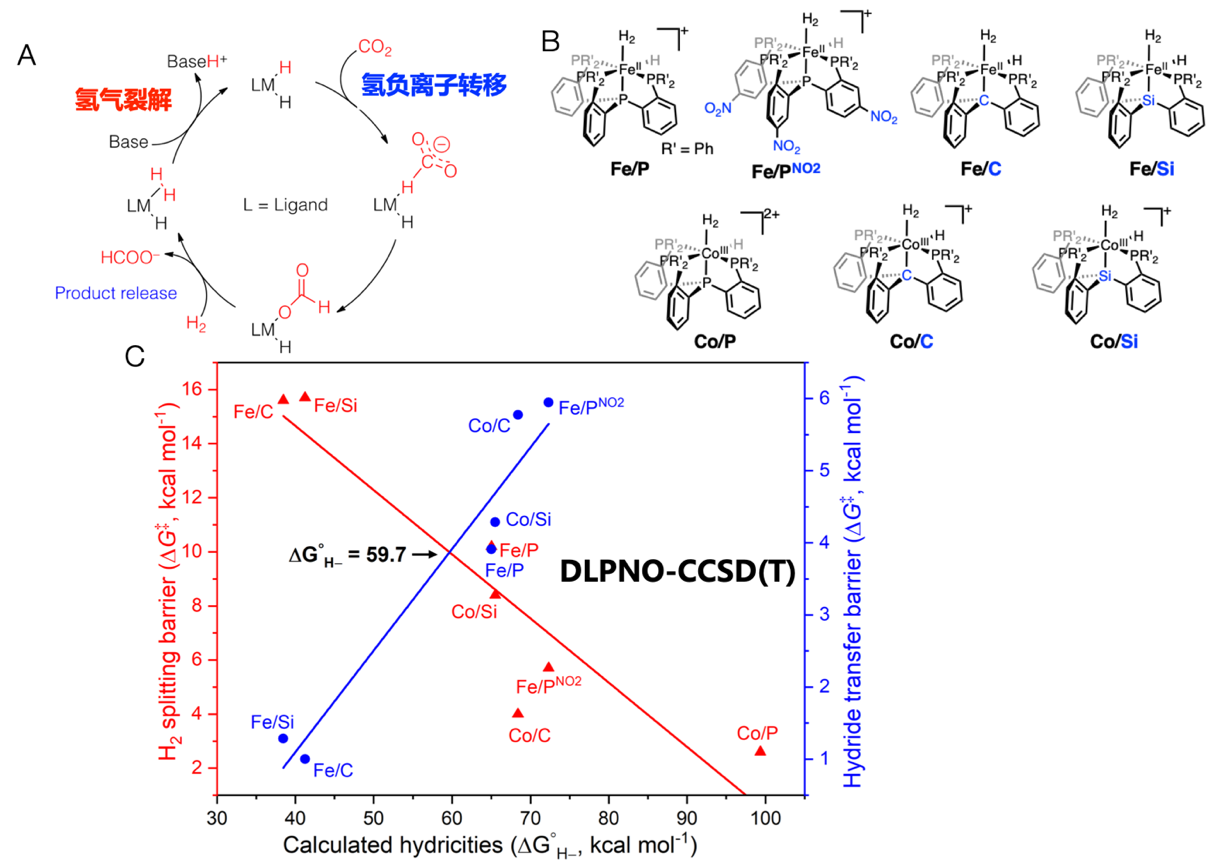
Our earlier computations studies using DFT and ab initio approaches proposed a step-wise single-electron reduction mechanism for O2 activation, namely, upon O2 binding induces one-electron transfer from to the Fe center to O2 leading to formation of a superoxide intermediate, which can convert into a peroxide with the co-substrate acting as the electron donor. Prior to O-O bond breaking to afford a high-valent Fe-oxo species, this peroxide complex can undergo one-electron reduction to yield a half-bond intermediate, the three-electron reduced form of O2 having a half O-O σ bond. Our recent experimental investigations using various spectroscopic techniques, most along with our synthetic collaborators, Prof. Dr. Way-Zen Lee at the Taiwan Normal University and Prof. Dr. Hua-Fen Hsu at National Cheng Kung University, provide strong support to the aforementioned mechanism. Specifically, treating divalent Fe, Mn and Co precursors with O2 at low temperatures, we detected generation of trivalent metal-superoxo complexes, which then can activate weak C-H and O-H bonds to yield the corresponding metal-hydroperoxo species. In the reaction of alkane oxidation by H2O2 catalyzed by low-spin ferric complexes, we found spectroscopic evidence for the existence of the half-bond intermediate. The high-valent Fe-oxo species was characterized to be the actual oxidant for the following substrate functionalization in a series of metalloenzymes.
Presently, we are working on discovery of novel chemical activity of the four different types of intermediates generated by step-wise O2 reduction, correlation of their electronic structures with the activity as well as the mechanism of the reverse reaction, i.e. diatomic O-O, N-O and N-N bond formation derived from high-valent metal-oxo and -nitrido complexes.
Short-chain alkane oxidation
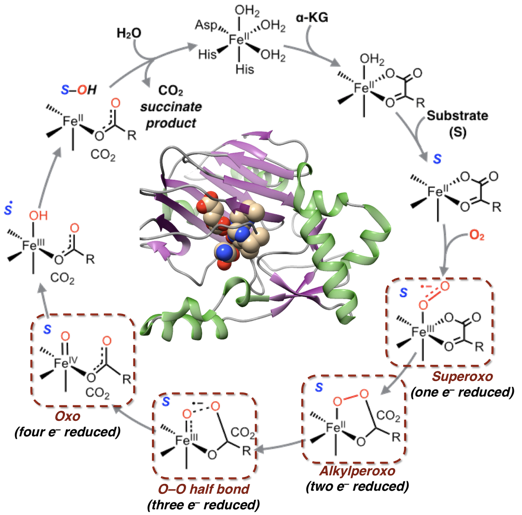
Our theoretical results suggest that most Fe(IV)-oxo biomimetics possess an S= 1 ground state with a low-lying S= 2 excited state. C-H bond oxidation can progress on the two potential energy surfaces; however, to complicate matters, there are two distinct σ and π pathways in the triplet and quintet reactions. In collaboration with Prof. Dr. Franc Meyer at the University of Göttingen, our recent work using MCD spectroscopy coupled to wavefunction based multireference CASSCF/NEVPT2 calculations revealed that the tetracarbene Fe(IV)-oxo species features a different electronic structure relative to those supported by neutral N donors. As a consequence, the C-H bond oxidation process with the former complex only occurs on the triplet surface. This finding indicated that the carbene complex is a mechanistically much simpler system allowing for experimentally probing the reaction mechanism, which we are pursuing.
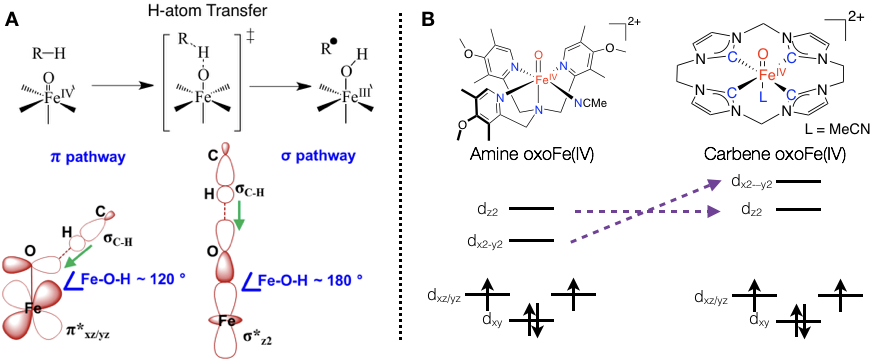
CO2 hydrogenation
The vast majority of catalysts for CO2 hydrogenation to yield formic acid often contain noble metals, such as Ru, Rh, Ir, etc, and those based on base metals often exhibit much lower activity. It was found that the reaction involves two key steps, namely, H2 splitting and hydride transfer and either of which can be the rate-liming step of the entire reaction. As shown in the well-accepted mechanism, in the H2-splitting step a metal-hydride bond is formed, whereas this bond is broken in the subsequent hydride-transfer step. Thus, we reasoned that the metal-hydride (M-H) bond strength, may be a key factor that dictates the barriers of both element process. To test this notion, we designed a series of Fe and Co complexes with different M-H bond strengths as quantified by its hydricity and estimated their catalytic activity using the highly corelated DLPNO-CCSD(T) approach. Our results showed that as the M-H bond strength increases, the barrier for H2 splitting drops; by contrast, the barrier for hydride transfer rises. Consequently, the systems near the crossing point, Fe/P and Co/Si, are likely to strive a delicate balance between the opposite requirement of the two step, and hence are probably good catalysts. Experimental, system Fe/P has been indeed shown to have superior activity of all non-noble CO2-hydrogenation catalysts reported thus far. Further experimental work is being carrying out to test the proposed catalyst-design strategy.