
Bioinorganic catalysis
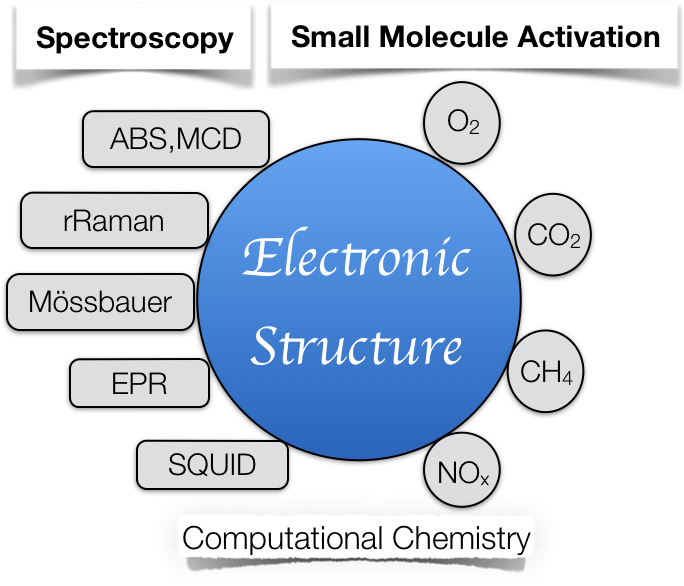
To tackle the current global energy crisis that human being is being confronted with, activation of energy-relevant small molecules, such as N2, O2, H2O, CO2, etc, attracts much attention and becomes a hot topic for a large body of research work. In this regard, nature has developed a wide range of highly efficient catalysts, enzymes, the majority of which contain transition metal centers. Typically, enzymatic reactions are found to proceed with unrivalled efficiency and exhibit remarkable, even exclusive in most cases, regio- and stereo-specificity. Undoubtedly, studies aimed at understanding the underlying mechanisms of such reactions, in particular, pinpointing pivotal features responsible for the found high efficacy and specificity are of fundamental significance. Chemical insights thus obtained can serve as a guide for tuning catalytic activity of available systems and, more importantly, trigger new ideas for designing novel catalysts with tailored properties.
Dissecting reaction mechanisms of small-molecule activation mediated by metalloenzymes and biomimetics has long been the central research topic of our group. Our working philosophy is that the prerequisite to elucidate the reaction mechanism is to analyze the electronic structures of kinetically competent intermediates, usually of fleeting nature, in the catalytic cycle. As such, detailed spectroscopic characterizations are often superior choices than crystallographic analyses, in light of the short lifetime of the species of interest. To do so, electron paramagnetic resonance (EPR), 57Fe Mössbauer, magnetic circular dichroism (MCD), resonance Raman (rR) and infrared (Ir) spectroscopies are widely employed techniques in our group. Our research work first focuses on identifying the characteristic spectroscopic signatures of important intermediates, which allows us to detect such transient species in actual catalytic cycles, and then correlates the electronic structures with the observed catalytic activity.
To probe the reaction mechanism, we also apply quantum chemical calculations using standard density functional theory (DFT) and highly correlated wavefunction based single- (CCSD(T)) and multi-reference (CASSCF/NEVPT2) approaches. The purpose of carrying out computational studies is not only to predict reliable reaction energies and barrier heights, but also to provide understanding of chemical transformations and ultimately set up the structure-activity relationship of key intermediates. Moreover, highly accurate calculations likely give us hints about what intermediate can be trapped and hence direct our further spectroscopic investigations. In parallel, prediction of spectroscopic parameters by first principle computations can aid in interpreting our experimental data and thus determining the nature of the intermediates.

