本研究组采用实验与理论计算结合,长期研究分子氧活化机理、低碳烷烃碳氢键的氧化机理、CO2加氢还原反应机理。
金属酶和模拟配合物活化分子氧
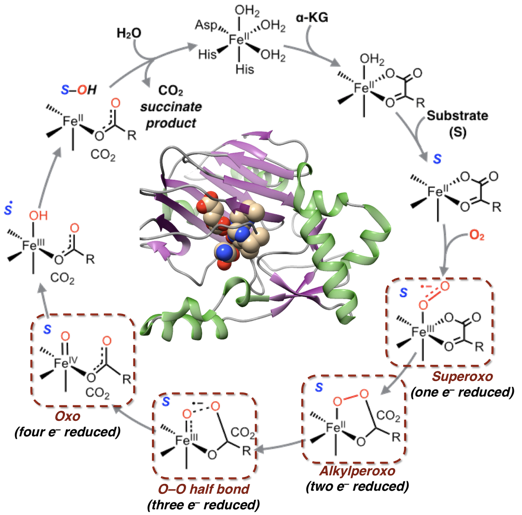
本研究组采用精确的CASSCF/NEVPT2的量子化学方法研究了一类单核非血红素含铁金属酶活化分子氧的反应机理。从头算(ab initio)和密度泛函DFT计算表明,分子氧活化经历逐步的单电子还原过程。其中关键步骤是铁中心和分子氧结合生成超氧配合物,其次是过氧配合物发生金属到过氧键的单电子转移生成分子氧三电子还原产物。在O-O键断裂形成高价铁氧化物之前,过氧化物经历单电子还原形成一个半键中间体,分子氧三电子还原产物中有半个O-Oσ键。台湾国立师范大学的李位仁教授和台湾国立成功大学的许铧芬教授基于上面的反应机理,采用多种光谱学技术进行了实验论证。实验表明低温下二价Fe,Mn,Co与分子氧反应生成相应的三价金属超氧配合物,可以活化C-H键和O-H键生成三价金属过氧化氢配合物。在低自旋的三价铁模拟配合物催化H2O2,进而氧化烷烃C-H键的反应中,本研究组通过多种光谱特征证明了半键中间体的存在。在一系列金属酶的催化反应中,形成的高价铁氧配合物可以作为下一步的氧化剂进行反应。
目前,本研究组致力于研究分子氧逐步还原下产生的四种中间体的化学活性,电子结构与活性之间的关系以及逆反应机理,即双原子分子O-O,N-O,N-N键的合成来自于高价金属的氧或氮配合物。
氧化低碳烷烃碳氢键
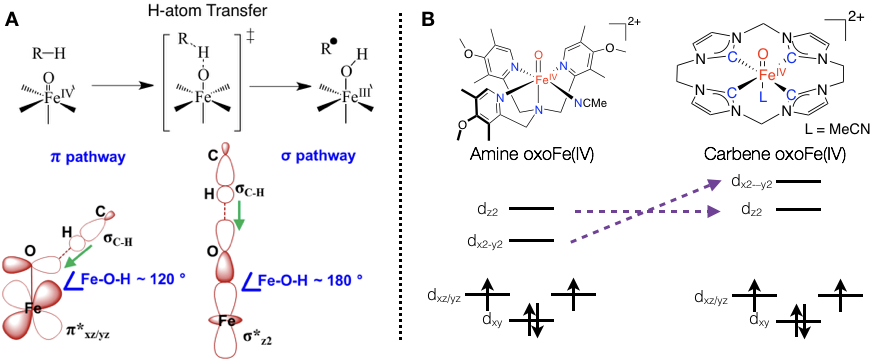
本研究组用从头算的CASSCF/NEVPT2方法预测四价铁端基氧模拟配合物一般都具有三重态基态和一个能量很近的五重激发态;碳氢键活化可以同时在三重态或五重态势能面上进行,而且在每一个势能面上都只有两条可能的氢原子转移的反应途径σ和π,我们与Franc Meyer教授合作,采用磁圆二色谱与CASSCF/NEVPT2理论计算相结合,首次在实验上证明中性氮配体的四价铁端基氧配合物确实具有三重态基态和一个能量很近的五重激发态(<3 kcal/mol),而卡宾配合物的五重态能量很高(>18 kcal/mol),因此后者活化碳氢键只能在三重态势能面上进行,这一发现简化了卡宾配合物活化碳氢键的反应机理。
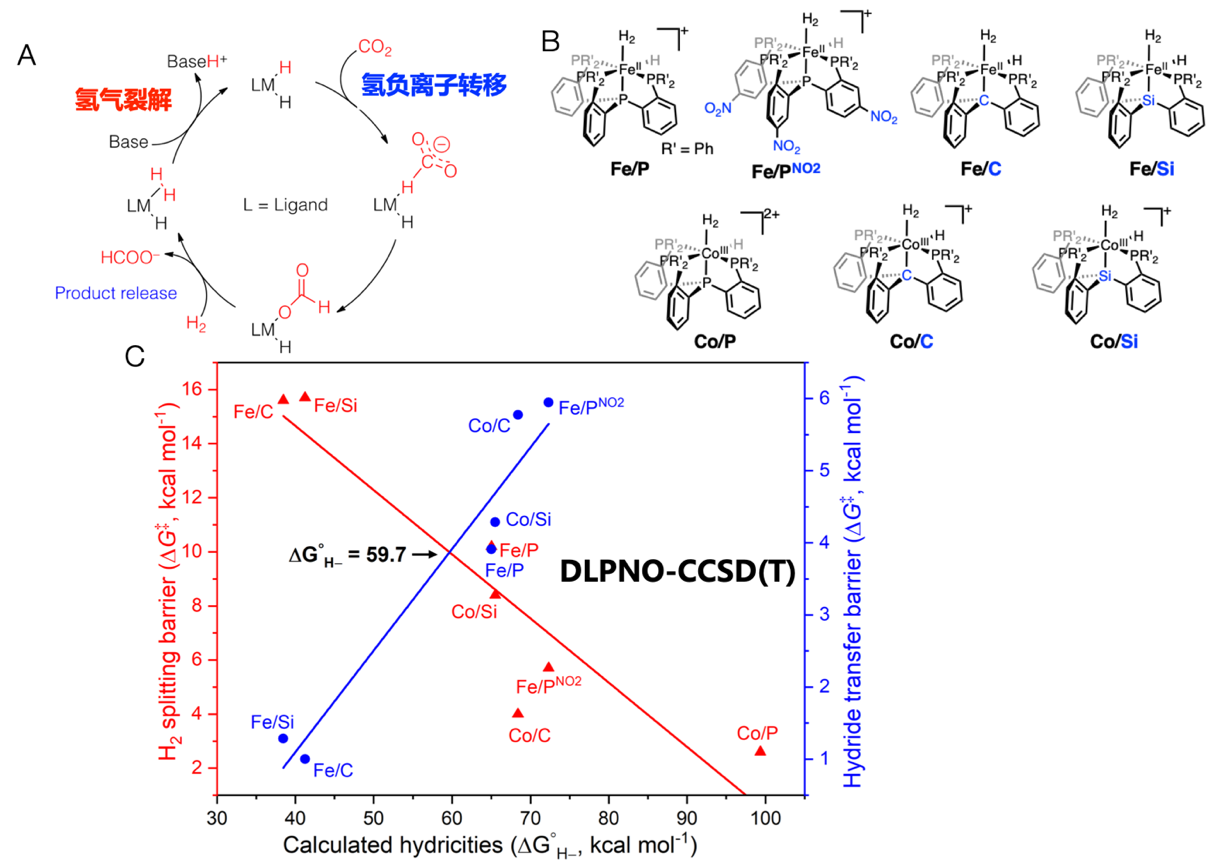
CO2加氢还原
现今报道的二氧化碳加氢转化为甲酸的催化剂绝大多数含有铑、铱等贵金属,而且个别报道的贱金属铁、钴催化剂的效率都显著低于贵金属催化剂。该反应有两个关键步骤:氢气裂解和氢负离子的转移,其中有一个是决速步骤。图A所示的反应机制中,氢气裂解形成了金属氢化物(M-H),在氢负离子转移这一步中断开。我们用从头算的DLPNO-CCSD(T)方法研究其反应机理。计算结果表明金属氢化物中金属中心和氢负离子化学键(M-H–)的强弱控制这两个关键步骤的反应势垒。M-H–键的强弱可用氢负离子活性(ΔG°H–)来定量描述。在氢气裂解中,因为形成金属-氢负离子键,氢负离子活性与反应势垒呈反比。而在氢负离子转移中,这个键需要断裂,所以氢负离子活性与反应势垒呈正比。这样的分析得出处于交叉点附近的Fe/P和Co/Si是高性能的催化剂。Fe/P体系是Mattias Beller组已经报道的高效贱金属催化剂。Jonas Peters课题组对Co/Si体系进行了实验研究,验证了其优异的二氧化碳加氢性能(J. Am. Chem. Soc. 2019, 141, 2814;Inorg. Chem. 2016, 55, 5438;Inorg. Chem. 2015, 54, 7192;Curr. Opin. Chem. Biol. 2015, 25, 103.)。